New: RNA Virtual Torsion Plot Service
EMDR Note: this service has been discontinued as of 2026
EMDR has a new web service for validation of RNA structures determined using cryo-EM and other experimental methods. RNA adopts complex three-dimensional folding patterns in biology such as ribosomal particles, spliceosomes, RNA viruses, and polymerase complexes. Virtual torsion angle plots provide a means to analyse RNA conformation in a manner analogous to the Ramachandran plot for proteins.1,2
The new service evaluates single structures, either by file upload or by entering a PDB id. Three flavors of pseudotorsion dihedral angle pairs are calculated for each non-terminal RNA residue using DSSR3,4. Coordinates are taken from phosphate atom positions, alternating with either C4' atom (η,θ, as shown below), C1' atom (η',θ'), or DSSR's base origin positions (η",θ").
Measured torsion/dihedral angle pairs are compared against the statistical distribution of a benchmark set of high quality, non-redundant5 RNA structures determined by X-ray crystallography at ≤ 2.5 Å. Separate analyses are performed for nucleotides with C3'-endo and C2'-endo ribose sugar conformations. A table of outliers is provided. Outliers can be legitimate but less-common backbone conformations, or they may indicate modelling error.
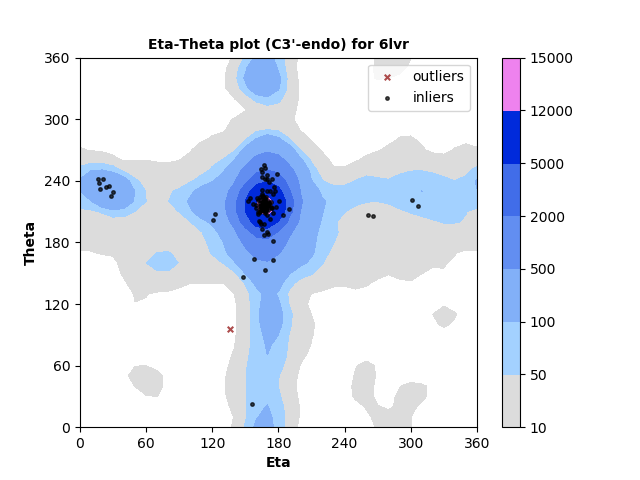
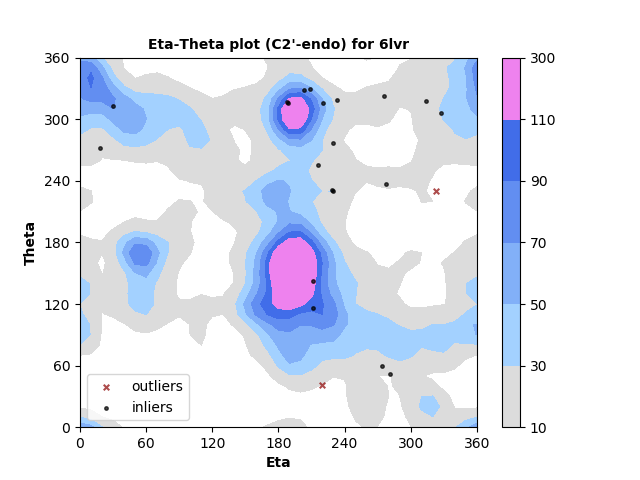
RNA residues with C3'-endo sugar pucker are plotted at left and with C2'-endo sugar pucker are plotted right. Each point represents the dihedral torsion angle pair eta (C4'n-1-Pn-C4'n-Pn+1, x-axis) vs. theta (Pn-C4'n-Pn+1-C4'n+1, y-axis) for RNA residue n. In this example most residues are in well-populated regions with just a small number identified as outliers. Outliers can either indicate modeling errors or rare conformations. They are identified in a table so that they can be further inspected.
Server home page: https://ptp.emdataresource.org. Navigate directly to results for a specific PDB entry (e.g., 6lvr): https://ptp.emdataresource.org/ptp/pdbid=6lvr.
- Wadley, L. M., Keating, K. S., Duarte, C. M. & Pyle, A. M. Evaluating and learning from RNA pseudotorsional space: quantitative validation of a reduced representation for RNA structure. J Mol Biol 372, 942-957, doi (2007).
- Keating, K. S., Humphris, E. L. & Pyle, A. M. A new way to see RNA. Q Rev Biophys 44, 433-466, doi (2011).
- Lu, X. J., Bussemaker, H. J. & Olson, W. K. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res 43, e142, doi (2015).
- Li, S., Olson, W. K., Lu, X. J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Research 47, W26-W34 (2019). doi
- Leontis, N. B., & Zirbel, C. L. Nonredundant 3D Structure Datasets for RNA Knowledge Extraction and Benchmarking. In RNA 3D Structure Analysis and Prediction, N. Leontis & E. Westhof (Eds.), (Vol. 27, pp. 281-298). Springer Berlin Heidelberg. doi (2012).